MD Simulationen
Obwohl experimentelle Methoden unumgänglich sind, um das Verhalten von Biomakromolekülen zu entschlüsseln, fehlen ihnen meist atomare Details. Hier sind Molekulardynamik-Simulationen (MD-Simulationen) die Methode der Wahl, um die experimentellen Ergebnisse zu ergänzen. Aufgrund der Größe der untersuchten Moleküle sind unsere "Arbeitspferde" Programme, die Newton'sche Mechanik mit klassischen Potentialen für die Kerne einsetzen, in speziellen Fällen aber auch quantenmechanische Methoden sowie kombinierte quantenmechanische und molekularmechanische (QM/MM) Programme und Monte Carlo (MC) Simulationen.
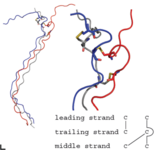
Ein Beispiel für die Anwendung der rein klassischen Molekulardynamik ist die Untersuchung einer cysteinreichen Kollagenregion. Für dieses große Protein wurde eine Reihe verschiedener Modelle simuliert, um mehr Informationen über seine mögliche Struktur zu erhalten (siehe Abb. 1 rechts, entnommen von Ref. 1).
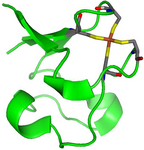
Ein Fall, bei dem ein QM/MM-Ansatz gewählt wurde, ist Rubredoxin, ein Enzym mit einem katalytisch aktiven Eisen-Schwefel-Zentrum, das in einem quantenmechanischen Rahmen beschrieben werden musste, während der verbleibende Teil des Proteins mit einer kostengünstigeren Technik behandelt wurde (siehe Abb. 2 und 3, entnommen aus Ref. 2).
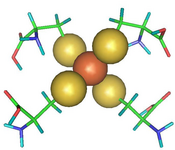
Da Kooperativität für die Faltung und Funktion von Proteinen von zentraler Bedeutung ist, sind wir generell sehr interessiert an diesem Thema. Kleine (im Vergleich zu Bio-Molekülen) Modellsysteme, die es erlauben, die Kooperativität zu untersuchen, sind Spin-Crossover-Verbindungen. In einer kürzlich durchgeführten Studie haben wir einen quantenmechanischen Ansatz verwendet, um zum Verständnis der Kooperativität in diesen Verbindungen beizutragen.

Hauke Paulsen

Gebäude 61
,
Raum 221
paulsen(at)physik.uni-luebeck.de
+49 451 3101 4203